The use of genome sequences from strains authenticated to appropriate species degree is a prerequisite for confidently exploring the evolutionary relationship amongst associated species. Aspergillus strains erroneously curated as Aspergillus oryzae and Aspergillus fumigatus have been seen in the National Center for Biotechnology Information (NCBI) genome database. Aspergillus parasiticus is one of a number of aspergilli that produce aflatoxin, the most potent carcinogenic mycotoxin recognized to this point. To make sure that legitimate conclusions are drawn by researchers from their genomics-related research, molecular analyses had been carried out to authenticate identities of A. parasiticus strains in the NCBI genome database.
Two of the 9 supposedly A. parasiticus strains, E1365 and NRRL2999, had been discovered to be misidentified. They turned out to be Aspergillus flavus based mostly on genome-wide single nucleotide polymorphisms (SNPs) and genetic options related to manufacturing of aflatoxin and cyclopiazonic acid. NRRL2999 lacked the further partial aflatoxin gene cluster recognized to be current in its equal pressure, designated as SU-1, and shared a really low whole SNPs rely particularly with A. flavus NRRL3357 however not with different A. flavus isolates. Therefore, the mislabeled NRRL2999 pressure really is a clonal pressure of A. flavus NRRL3357, whose genome was first sequenced in 2005.
Mutation breeding is a unprecedented software in plant breeding to extend the genetic variability, the place mutations in anthocyanin biosynthesis are targets to generate distinctive phenotypes in decorative species. In poinsettia, ionizing radiation is routinely utilized in breeding packages to acquiring a variety of colors, with practically all pink and white varieties being obtained after γ- or X-ray mutagenesis of pink varieties. In the current research we carried out a radical characterization of a possible mutagenesis goal gene as the major accountable for the ‘white paradox’ in poinsettia. Moreover, overexpression of Bract1 wild-type allele in Arabidopsis tt19 mutants restored the anthocyanin phenotype, whereas the Bract1 mutated allele confirmed to be non-functional.
We recognized a GST gene in poinsettia (Bract1) as an important issue for the expression of anthocyanin-based pink colouration of bracts, which presents a excessive phylogenetic similarity to recognized anthocyanin-related GSTs. Red poinsettia varieties and white mutants generated from these varieties by X-ray had been analysed for polymorphisms associated to the ‘white paradox’ in the species. A Four bp mutation in a brief repeat inside the coding area of Bract1 is almost definitely accountable for the look of white phenotypes upon irradiation therapy. The polymorphism between wild-type and mutant alleles co-segregates with the phenotype in progeny from heterozygous pink and white mother and father.
Clinical and Molecular Attributes of Patients With BCR/ABL1-negative Myeloproliferative Neoplasms in India: Real-world Data and Challenges
Patients referred to Hematology-Oncology from January 2018 to August 2020 with suspected MPNs had been included in the evaluation and prospectively followed-up. All sufferers had been initially screened, and solely these assembly the up to date World Health Organization 2016 standards had been included in the evaluation. Epidemiologic, medical, and molecular traits had been documented, and sufferers had been followed-up prospectively. This is one of the first systematic descriptions and potential follow-up of sufferers with BCR/ABL-negative MPNs from India.
A complete of 233 sufferers had been referred for analysis of MPN, of which 63 had been included in the evaluation, together with 39 males and 24 females. The median age at analysis was 57 years (vary, 28-82 years), and 38% sufferers had been youthful than 50 years of age. The most typical shows had been incidental detection in 35 (55.5%), belly signs in 13 (20%), fatiguability in 7 (11%), and up to date vascular occasions in 6 (9.5%) sufferers. Final analysis was polycythemia vera in 27, important thrombocytosis (ET) in 21, prefibrotic myelofibrosis in 9, and myelofibrosis in 6 sufferers.
The frequency of driver mutations in polycythemia vera included JAK2 in 75%; in ET, JAK2 in 33%, CALR in 33%, and MPL in 4%; and in prefibrotic myelofibrosis, JAK2 in 66% and CALR in 33%. Aspirin was used for all sufferers together with risk-adapted cytoreduction with hydroxyurea. Ruxolitinib was reserved for signs refractory to hydroxyurea. After a median follow-up of 15 months (interquartile vary, 10-28 months) from analysis, illness development was famous in Four sufferers. Two sufferers died at the finish of the follow-up interval, together with 1 with secondary acute myeloid leukemia put up myelofibrosis and one with ET and coexistent oral malignancy. The remaining 61 sufferers are alive and on common therapy.
Genetic characterization of a novel HIV-1 second-generation recombinant type (CRF01_AE/07_BC) recognized in Yan`an metropolis, China
Due to co-epidemic of CRF01_AE and CRF07_BC in China, rising numbers of the second-generation recombinants between them have been recognized particularly amongst sexual inhabitants. Here, we recognized an distinctive CRF01_AE/CRF07_BC recombinants from a male HIV-1 postive particular person (18YA004) contaminated by heterosexual contact in Yan`an metropolis, Shaanxi province. The close to full-length genome analyses confirmed 18YA004 was divided into six fragments by 5 breakpoints situated in the pol, vpr, vpu and nef gene respectively.
Three CRF01_AE segments (phase Ⅰ, Ⅲ, Ⅴ) had been all clustered inside the cluster 4a lineage, completely circulating amongst MSM in the norther China. Coupled with our earlier discovering of CRF01_AE/C recombinant in Yan`an metropolis, the emergence of CRF01_AE/CRF07_BC pressure additional steered co-existence of mutiple HIV-1 genotypes right here. Therefore, it’s a necessity to proceed monitoring the molecular epidemiology of HIV-1 amongst high-risk teams to acquire a greater understanding of the genetic complexity and transmission of HIV-1 in this area.
The purpose of this work is to watch the state of the proteolytic group in time and house for the subsequent growth of approaches to an goal evaluation of the late postmortem interval. The examine proposes a mixture of customary bacterioscopic and bacteriological research strategies with strategies of molecular biology and genetics, which make it attainable to establish species and strains of mammalian corpses’ proteolytics at the degree of particular DNA or RNA. On the foundation of phenotypic traits and a comparative evaluation of the nucleotide sequences of genes encoding 16S rRNA, the species belonging of the remoted strains was proved.
The set of strategies’ mixture, together with conventional microbiological evaluation and molecular genetic research, appears promising each for the objective of substantiating and widespread use of microbiological strategies in forensic medical observe, and for growth an goal scientific base for establishing the cause-and-effect patterns of microbial transformation of natural matter in nature.Self-fertilization (additionally termed selfing) is a mode of replica that happens in hermaphrodites and has advanced a number of occasions in varied plant and animal species. A transition from outbreeding to selfing in hermaphroditic flowers is often related to modifications in flower morphology and performance.
This examine aimed to establish genetic results of selfing in the F2 progeny of F1 hybrid developed by crossing Lilium lancifolium with the Asiatic Lilium hybrid ‘Dreamland.’ Fluorescence in situ hybridization (FISH) and inter-simple sequence repeats (ISSR) methods have been used to detect genetic variations in crops produced by selfing. The FISH outcomes confirmed that F1 hybrid have been much like the feminine guardian (L. lancifolium) concerning the 45S loci, however F2 people confirmed variation in the quantity and placement of the respective loci. In F2 progeny, F2-2, F2-3, F2-4, F2-5, and F2-Eight hybrids expressed two robust and one weak 5S sign on chromosome 3, whereas F2-7 and F2-9 people expressed one robust and two weak alerts.
Only two robust 5S alerts have been detected in an F2-1 plant. The ISSR outcomes confirmed a most similarity worth of 0.6269 between the feminine guardian and the F2-2 hybrid. Regarding similarity to the male guardian, a most worth of 0.6119 was discovered in the F2-1 and F2-2 hybrids. The highest genetic distance from L. lancifolium and the Asiatic Lilium hybrid ‘Dreamland’ was noticed in the F2-Four progeny (0.6352 and 0.7547, respectively). Phylogenetic relationships confirmed that the F2 progeny have been nearer to the male guardian than to the feminine guardian. Self-fertilization confirmed results on variation amongst the F2 progeny, and results on the genome have been confirmed utilizing FISH and ISSR analyses.
Genetic and molecular biology of autism spectrum dysfunction amongst Middle East inhabitants: a assessment
Autism spectrum dysfunction (ASD) is a neurodevelopmental illness, characterised by impaired social communication, govt dysfunction, and irregular perceptual processing. It is extra frequent amongst males. All of these scientific manifestations are related to atypical neural growth. Various genetic and environmental danger components are concerned in the etiology of autism. Genetic evaluation is crucial for the early detection and intervention which might enhance social communications and scale back irregular behaviors. We have additionally categorized the reported genes based mostly on their cell and molecular features.
Although, there’s a noticeable ASD incidence in Middle East nations, there may be nonetheless a scarcity of information about the genetic and molecular biology of ASD amongst this inhabitants to introduce environment friendly diagnostic and prognostic strategies. In the current assessment, we have now summarized all of the genes which have been related to ASD development amongst Middle East inhabitants. This assessment clarifies the genetic and molecular biology of ASD amongst Middle East inhabitants and paves the means of introducing an environment friendly inhabitants based mostly panel of genetic markers for the early detection and administration of ASD in Middle East nations.
From mutation to mechanism: deciphering the molecular perform of genetic variants linked to human ageing
Many of the main causes of dying in people, equivalent to heart problems, kind 2 diabetes and Alzheimer’s illness are influenced by organic mechanisms that turn into dysregulated with growing age. Hence, by focusing on these ageing-related mechanisms, we might be able to enhance well being in outdated age. Ageing is partly heritable and genetic research have been reasonably profitable in figuring out genetic variants related to ageing-related phenotypes (lifespan, healthspan and longevity). To decipher the mechanisms by which the recognized variants affect ageing, research that focus on their useful validation are very important.
In this angle, we describe the steps that may very well be taken in the course of of useful validation: (1) in silico characterisation utilizing bioinformatic instruments; (2) in vitro characterisation utilizing cell strains or organoids; and (3) in vivo characterisation research utilizing mannequin organisms. For the in vivo characterisation, you will need to focus on translational phenotypes which are indicative of each healthspan and lifespan, equivalent to the frailty index, to tell subsequent intervention research. The depth of useful validation of a genetic variant relies upon on its location in the genome and conservation in mannequin organisms.
Moreover, some variants could show to be exhausting to characterise attributable to context-dependent results associated to the experimental setting or genetic background. Future efforts to functionally characterise the (newly) recognized genetic variants ought to shed mild on the mechanisms underlying ageing and can assist in the design of focused interventions to enhance well being in outdated age.
Sub-Saharan Africa was traditionally thought-about an animal influenza chilly spot, with solely sporadic extremely pathogenic H5 outbreaks detected over the past 20 years. However, in 2017, low pathogenic avian influenza A(H9N2) viruses had been detected in poultry in Sub-Saharan Africa. Molecular, phylogenetic, and antigenic characterization of isolates from Benin, Togo, and Uganda confirmed that they belonged to the G1 lineage. Isolates from Benin and Togo clustered with viruses beforehand described in Western Africa, whereas viruses from Uganda had been genetically distant and clustered with viruses from the Middle East. Viruses from Benin exhibited decreased cross-reactivity with these from Togo and Uganda, suggesting antigenic drift related to diminished replication in Calu-Three cells.
The viruses exhibited mammalian adaptation markers just like these of the human strainCigarette smoking is a serious threat issue for lung most cancers improvement and development; nevertheless, the mechanism of how cigarette smoke prompts signaling pathways in selling most cancers malignancy stays to be established. Herein, we aimed to find out the contribution of a signaling protein, myristoylated alanine-rich C kinase substrate (MARCKS), in smoke-mediated lung most cancers. We firstly examined the degrees of phosphorylated MARCKS (phospho-MARCKS) in smoke-exposed human lung most cancers cells and specimens in addition to non-human primate airway epithelium.
Next, the MARCKS-interactome and its gene networks had been recognized. We additionally used genetic and pharmacological approaches to confirm the performance and molecular mechanism of smoke-induced phospho-MARCKS. We noticed that MARCKS turns into activated in airway epithelium and lung most cancers cells in response to cigarette smoke. Functional proteomics revealed MARCKS protein instantly binds to NF-κB-activating protein (NKAP). Following MARCKS phosphorylation at ser159 and ser163, the MARCKS-NKAP interplay was inhibited, resulting in the activation of NF-κB signaling.
In a display of two cohorts of lung most cancers sufferers, we confirmed that phospho-MARCKS is positively correlated with phospho-NF-κB (phospho-p65), and poor survival. Surprisingly, smoke-induced phospho-MARCKS upregulated the expression of pro-inflammatory cytokines, epithelial-mesenchymal transition, and stem-like properties. Conversely, focusing on of MARCKS phosphorylation with MPS peptide, a selected MARCKS phosphorylation inhibitor, suppressed smoke-mediated NF-κB signaling exercise, pro-inflammatory cytokines expression, aggressiveness and stemness of lung most cancers cells. Our outcomes recommend that phospho-MARCKS is a novel NF-kB activator in smoke-mediated lung most cancers development and present a promising molecular mannequin for growing new anticancer methods.
Rapid choice response to ethanol in Saccharomyces eubayanus emulates the domestication course of beneath brewing circumstances
Although the everyday genomic and phenotypic adjustments that characterize the evolution of organisms beneath the human domestication syndrome symbolize textbook examples of fast evolution, the molecular processes that underpin such adjustments are nonetheless poorly understood. Domesticated yeasts for brewing, the place quick technology instances and giant phenotypic and genomic plasticity had been attained in just a few generations beneath choice, are prime examples. To experimentally emulate the lager yeast domestication course of, we created a genetically advanced (panmictic) synthetic inhabitants of a number of Saccharomyces eubayanus genotypes, one of the mother and father of lager yeast.
Then, we imposed a relentless choice regime beneath a excessive ethanol focus in 10 replicated populations throughout 260 generations (6 months) and in contrast them with propagated controls uncovered solely to glucose. Propagated populations exhibited a variety differential of 60% in progress fee in ethanol, largely defined by the proliferation of a single lineage (CL248.1) that competitively displaced all different clones. Interestingly, the result doesn’t require your complete time-course of adaptation, as 4 lineages monopolized the tradition at technology 120. Sequencing demonstrated that de novo genetic variants had been produced in all propagated strains, together with SNPs, aneuploidies, INDELs and translocations.
In addition, the completely different propagated populations confirmed correlated responses resembling the domestication syndrome: genomic rearrangements, quicker fermentation charges, decrease manufacturing of phenolic off-flavours and decrease unstable compound complexity. Expression profiling in beer wort revealed altered expression ranges of genes associated to methionine metabolism, flocculation, stress tolerance and diauxic shift, doubtless contributing to increased ethanol and fermentation stress tolerance in the advanced populations. Our examine reveals that experimental evolution can rebuild the brewing domestication course of in ‘quick movement’ in wild yeast, and additionally gives a robust software for learning the genetics of the variation course of in advanced populations.
The mitogenome of Ophidascaris wangi remoted from snakes in China
Different species of the genus Ophidascaris (Baylis, 1921; Nematoda: Ascaridida, Ascaridoidea) are intestinal parasites of varied snake species. More than 30 Ophidascaris species have been reported worldwide; nevertheless, few moleculargenetic research have been performed on this genus. We sequenced the whole mitogenome of Ophidascaris wangi parasitizing two snake species of the household Colubridae, i.e., Elaphe carinata (Günther, 1864) and Dinodon rufozonatum. The mitogenome sequence of O. wangi was roughly 14,660 base pairs (bp) lengthy and encoded 36 genes, together with 12 protein-coding genes (PCGs), 2 ribosomal RNA (rRNA) genes, and 22 switch RNA genes.
Gene association, genome content material, and transcription route had been in line with these in Toxascaris leonina (Linstow, 1902; Ascaridida: Ascarididae). Phylogenetics of O. wangi and different ascaridoids had been reconstructed based mostly on the concatenated amino acid sequences of 12 PCGs, and on nucleotide sequences of 12 PCGs and two rRNA genes. Phylogenetic analyses had been carried out utilizing most probability and Bayesian inference strategies, and the outcomes prompt that O. wangi constitutes a sister clade of Ascaris, Parascaris, Baylisascaris, and Toxascaris inside the household Ascarididae, which is a sister clade of Toxocaridae.
The mitogenome sequence of O. wangi obtained from the current examine will probably be helpful for future identification of the nematode worms in the genus Ophidascaris and will enhance the understanding of inhabitants genetics, molecular epidemiology, and phylogenetics of ascaridoid nematodes in snakes.
<i>Pseudomonas syringae</i> pv. <i>phaseolicola</i> (P.s. phaseolicola) is one of about 45 acknowledged pathovars inside the <i>P. syringae</i> group and is the causal agent of halo-blight illness of beans. DNA from this bacterium digested to completion with two totally different restriction enzymes, <i>Pac</i>I and <i>Pme</i>I, yielded 15 and 16 fragments, respectively. These have been separated utilizing PFGE and sized by comparability to recognized <em>molecular</em> mass markers. The <i>P.s. phaseolicola</i> chromosome was decided to be roughly 5.64 Mb in dimension.
To hyperlink the totally different fragments obtained right into a round chromosome map for each enzymes, 150 random Tn<i>5</i> mutants of <i>P.s. phaseolicola</i> have been used as a supply of DNA and the identification of the band carrying the transposon ‘tag’ in every mutant was accomplished after PFGE and Southern hybridization of an entire chromosomal digestion utilizing a Tn<i>5</i> probe. Partial digestions of DNA from totally different Tn<i>5</i> mutants ‘tagging’ particular bands have been then generated and the full and partial merchandise of the digestion separated by PFGE and recognized with a Tn<i>5</i> probe.
By calculating the dimension of the partial merchandise, it was then attainable to hyperlink totally different bands right into a bodily map. This is the first report on the development of a bodily map of a member of the P. syringae group and needs to be invaluable for <em>molecular</em> <em>genetic</em> evaluation on this species and in evolutionary or taxonomic research when in comparison with comparable information obtained for any of the different acknowledged pathovars. In Caulobacter crescentus, this nanofilament, although essential for floor colonization, has by no means been completely investigated at the molecular degree.
Bacterial pili are proteinaceous motorized nanomachines that play numerous practical roles together with floor adherence, bacterial movement, and virulence. The surface-contact sensor kind IVc (or Tad) pilus is broadly distributed in each Gram-positive and Gram-negative micro organism. As Caulobacter assembles a number of floor appendages at particular phases of the cell cycle, we designed a fluorescence-based display screen to selectively research single piliated cells and mixed it with atomic pressure microscopy and genetic manipulation to quantify the nanoscale adhesion of the kind IVc pilus to hydrophobic substrates.
Deep studying approaches for pure product discovery from plant endophytic microbiomes
Plant microbiomes should not solely numerous, but in addition seem to host an enormous pool of secondary metabolites holding nice promise for bioactive pure merchandise and drug discovery. Yet, most microbes inside crops look like uncultivable, and for these that may be cultivated, their metabolic potential lies largely hidden by regulatory silencing of biosynthetic genes. The current explosion of highly effective interdisciplinary approaches, together with multi-omics strategies to handle multi-trophic interactions and synthetic intelligence-based computational approaches to deduce distribution of operate, collectively current a paradigm shift in high-throughput approaches to pure product discovery from plant-associated microbes.
Arguably, the key to characterizing and harnessing this biochemical capability is determined by a novel, systematic method to characterize the triggers that activate secondary metabolite biosynthesis by molecular or genetic indicators from the host plant, members of the wealthy ‘in planta’ group, or from the setting. This assessment explores breakthrough approaches for pure product discovery from plant microbiomes, emphasizing the promise of deep studying as a software for endophyte bioprospecting, endophyte biochemical novelty prediction, and endophyte regulatory management.
It concludes with a proposed pipeline to harness international databases (genomic, metabolomic, regulomic, and chemical) to uncover and unsilence fascinating pure merchandise. In gentle of this rising understanding, the G. tritici-wheat interplay might present a mannequin research system for root-infecting fungal pathogens of cereals.
Take-All Disease: New Insights into an Important Wheat Root Pathogen
Take-all illness, attributable to the fungal root pathogen Gaeumannomyces tritici, is taken into account to be the most vital root illness of wheat worldwide. Here we assessment the advances in take-all analysis over the final 15 years, specializing in the identification of new sources of genetic resistance in wheat family members and the function of the microbiome in illness growth. We additionally spotlight current breakthroughs in the molecular interactions between G. tritici and wheat, together with genome and transcriptome analyses. These new findings will support the growth of novel management methods in opposition to take-all illness.
The growing demand for environment friendly and strong processes in the purification of monoclonal antibodies (mAbs) has not too long ago introduced frontal chromatography to the forefront. Applied throughout the sprucing step, it permits the elimination of excessive molecular weight aggregates from the goal product, reaching excessive purities. Typically, this course of is operated in batch utilizing a single column, which makes it intrinsically subjected to a purity-yield tradeoff. This implies that excessive purities can solely be achieved at the value of decreasing the product yield and vice versa.
Description: Intact Genomics Taq DNA Polymerase is a thermostable DNA polymerase that possesses a 5´→3´ polymerase activity (1, 2) and a 5´ flap endonuclease activity (3, 4). This product is supplied with 10x PCR reaction buffer, containing MgCl2, which produces a final Mg2+ concentration of 1.5 mM. Ideal for primary extension reaction with DNA fragments having dA overhang on 3’ ends.Product Includes:Taq DNA Polymerase10x PCR Buffer with Mg2+5x Magic Enhancer
Taq DNA Polymerase - 5,000 units with separate dNTP Mix
Description: Intact Genomics Taq DNA Polymerase is a thermostable DNA polymerase that possesses a 5´→3´ polymerase activity (1, 2) and a 5´ flap endonuclease activity (3, 4). This product is supplied with 10x PCR reaction buffer, containing MgCl2, which produces a final Mg2+ concentration of 1.5 mM. Ideal for primary extension reaction with DNA fragments having dA overhang on 3’ ends.Product Includes:Taq DNA Polymerase10x PCR Buffer with Mg2+5x Magic Enhancer10mM dNTP Mix
Recently, a two-column steady implementation of frontal chromatography, known as Flow2, was developed (Vogg et al., J. Chrom. A, 1619, 460943, 2020). Despite having the ability of assuaging the purity-yield tradeoff typical of batch operations, the enhance in the quantity of course of parameters complicates its optimum design, with the threat of not exploiting its full potential. In this work, we developed an advert hoc design process appropriate for the optimization of each batch frontal chromatography and Flow2 in phrases of purity, yield and productiveness. This process supplied comparable outcomes as a multi-objective optimization primarily based on genetic algorithm however with decrease computational effort.
Climate change globally perturbs water circulation thereby influencing ecosystems together with cultivated land. Both dangerous and useful species of bugs are prone to be susceptible to such adjustments in local weather. As small animals with a disadvantageous floor space to physique mass ratio, they face a danger of desiccation. A quantity of behavioural, physiological and genetic methods are deployed to unravel these issues throughout adaptation in varied Drosophila species. Over 100 desiccation-related genes have been recognized in laboratory and wild populations of the cosmopolitan fruit fly Drosophila melanogaster and its sister species in large-scale and single-gene approaches.
These genes are concerned in water sensing and homeostasis, and barrier formation and performance through the manufacturing and composition of floor lipids and through pigmentation. Interestingly, the genetictechnique applied in a given inhabitants seems to be unpredictable. In half, this can be as a consequence of completely different experimental approaches in completely different research. The noticed variability might also replicate a wealthy standing genetic variation in Drosophila permitting a quasi-random selection of response methods by soft-sweep occasions, though additional research are wanted to unravel any underlying ideas.
These findings underline that D. melanogaster is a sturdy species properly tailored to withstand local weather change-related desiccation. The wealthy knowledge obtained in Drosophila analysis present a framework to handle and perceive desiccation resistance in different bugs. Through the applying of highly effective genetic instruments in the mannequin organism D. melanogaster, the capabilities of desiccation-related genes revealed by correlative research might be examined and the underlying molecular mechanisms of desiccation tolerance understood. The mixture of the wealth of obtainable knowledge and its genetic accessibility makes
Drosophila a great bioindicator. Accumulation of knowledge on desiccation resistance in Drosophila might permit us to create a world map of genetic evolution in response to local weather change in an insect genome. Ultimately these efforts might present tips for coping with the consequences of climate-related perturbations on insect inhabitants dynamics in the long run. In the brand new period of genetic profiling of tumors and focused therapeutics, this assessment describes the epidemiology, pathology, molecular traits, and present administration with ongoing medical trials for chRCC.
Chromophobe renal cell carcinoma (chRCC) is the third most typical sort of RCC with distinct biology in comparison with different kidney most cancers subtypes. The heterogeneity between the RCC subtypes is related to noticeable variations in tumor aggressiveness and danger for the event of metastatic illness. ChRCC is characterised by chromosomal aneuploidy, TP53, PTEN, and mitochondrial gene mutations. Though the therapeutic panorama of clear cell RCC (ccRCC) has considerably advanced over the previous decade, restricted progress has been seen in chRCC as a consequence of its rare incidence. In truth, the therapeutic method for chRCC is usually extrapolated from ccRCC therapies or research that mix a number of varieties of nccRCC subtypes.
Identification and characterization of key haem pathway genes related to the synthesis of porphyrin in Pacific oyster (Crassostrea gigas)
Molluscs exhibit numerous shell colours. The molecular regulation of shell coloration is nonetheless not properly understood. To examine the connection of shell coloration with pigment synthesis, we analyzed the distribution of porphyrins, a widespread group of pigments in nature, in 4 Pacific oyster strains of completely different shell colours together with black, orange, golden, and white. The porphyrin distribution was analyzed in oyster mantles and shells by fluorescence imaging and UV spectrophotometer. The outcomes confirmed that crimson fluorescence emitted by porphyrins below the UV gentle was detected solely on the nacre of the orange-shell pressure and mantles of orange, black and white-shell strains.
Extracts from newly deposit shell, nacre and mantle tissue from orange-shell specimens confirmed peaks in UV-vis spectra which might be attribute of porphyrins, however these weren’t noticed for the opposite shell-color strains. In addition, genes of the haem artificial pathway have been remoted and characterised. Phylogenetic evaluation of CgALAS, CgALAD, CgPBGD, CgUROS, and CgUROD present additional proof for a conserved genetic pathway of haem synthesis throughout evolution. Differential expression of the haem genes expressed in mantle tissues assist these findings and are according to porphyrins being produced by the orange pressure solely.
Tissue in situ hybridization demonstrated the expression of these candidate genes on the outer fold of C. gigas mantles the place shell is deposited. Our research present a greater understanding of shell pigmentation in C. gigas and candidate genes for future mechanistic evaluation of shell coloration formation in molluscs. A excessive intraspecific genetic range was noticed in the echinostomatid, notocotylid, echinochasmid, and heterophyid species, whose definitive hosts embrace birds.
Trematode range in freshwater snails from a stopover level for migratory waterfowls in Hokkaido, Japan: An evaluation by molecular phylogenetic and inhabitants genetic analyses
The cryptic range of trematodes was evaluated in the Nagayama-shinkawa River, a man-made canal of the Ishikari River System of Hokkaido, Japan. Numerous migratory waterfowls use the canal as a stopover level in each spring season. The lymnaeid snail, Radix auricularia, and the semisulcospirid snail, Semisulcospira libertina, colonize the static and flowing water areas, respectively. The trematode fauna of the 2 snails was assessed by molecular phylogenetic and inhabitants genetic analyses. Each of distinctive clades in mitochondrial DNA bushes was arbitrarily set as a species.
In whole, 14 species of the households Diplostomidae, Echinostomatidae, Notocotylidae, Plagiorchiidae, and Strigeidae occurred in R. auricularia, wherease S. libertina harbored 10 species of the households Echinochasmidae, Heterophyidae, Notocotylidae, and Lecithodendridae and Cercaria creta, an unclassified species whose grownup stage remains to be unknown. The species range of the larval trematodes may very well be acknowledged as a “scorching spot”, suggesting that the seasonal go to of waterfowls is essential to unfold trematodes and to maintain their range. It appears seemingly that every of the parasite populations is at all times disturbed by repeated visits of waterfowls.
Recently, it was discovered that proteomes from poliovirus, measles virus, dengue virus, and extreme acute respiratory syndrome-related Coronavirus 2 (SARS-CoV-2) have excessive molecular mimicry on the heptapeptide degree with the human proteome, whereas heptapeptide commonality is minimal or absent with proteomes from nonhuman primates, that’s, gorilla, chimpanzee, and rhesus macaque. To purchase extra information on the problem, analyses right here have been expanded to Ebola virus, Francisella tularensis , human immunodeficiency virus-1 (HIV-1), Toxoplasma gondii , Variola virus, and Yersinia pestis . Results affirm that heptapeptide overlap is excessive between pathogens and Homo sapiens , however not between pathogens and primates. Data are mentioned in mild of the doable genetic bases that otherwise mannequin primate phenomes, thus presumably underlying the zero/low degree of molecular mimicry between infectious brokers and primates.
The Genome Reference Consortium (GRC) has in line with its personal assertion the “mission to enhance the human reference genome meeting, correcting errors and including sequence to make sure it offers the most effective illustration of the human genome to satisfy fundamental and medical analysis wants”. Data from GRC is included in genome browsers like UCSC (University of California, Santa Cruz), Ensembl or NCBI (National Center for Biotechnology Information) and are thereby bases for scientific and diagnostically working human genetic neighborhood. There have been three main factors recognized: (1) GRC missed to together with three chromosomal subbands, every, for 1q32.1, 2p21, 5q13.2, 6p22.3 and 6q21, which have been outlined by International System for Human Cytogenetic Nomenclature (ISCN) already again in 1980s
as a substitute GRC included extra 6 subbands not ever acknowledged by ISCN. (2) GRC outlined 34 chromosomal subbands of 0.1 to 0.9 Mb in measurement, whereas it’s normal settlement of cytogeneticists that it unlikely to detect chromosomal aberrations under 1-2 Mb in measurement by GTG-banding. And (3): nonetheless all sequences utilized in molecular cytogenetic routine diagnostics to detect heterochromatic and/ or pericentromeric satellite tv for pc DNA sequences throughout the human genome will not be included but into human reference genome. For these sequences, localization and approximate sizes have been decided within the 1970s to 1990, and if included at the very least ~ 100 Mb of the human genome sequence might be added to the genome browsers.
Current progress in medical, molecular, and genetic elements of grownup fibromuscular dysplasia
Fibromuscular dysplasia (FMD) is a non-atherosclerotic vascular illness that will contain medium-sized muscular arteries all through the physique. The majority of FMD sufferers are ladies. Although a spread of genetic, mechanical, and hormonal components play a task within the pathogenesis of FMD, total, its trigger stays poorly understood. It is possible that the pathogenesis of FMD is linked to a mixture of genetic and environmental components. Extensive research have correlated the arterial lesions of FMD to histopathological findings of arterial fibrosis, mobile hyperplasia, and distortion of the irregular structure of the arterial wall. More lately, the vascular phenotype of lesions related to FMD has been expanded to incorporate arterial aneurysms, dissections, and tortuosity.
However, within the absence of a string of beads or focal stenosis, these lesions don’t suffice to ascertain the analysis. While FMD mostly entails renal and cerebrovascular arteries, involvement of most arteries all through the physique has been reported. Increasing proof highlights that FMD is a systemic arterial illness and that subclinical alterations will be present in non-affected arterial segments. Recent vital progress in FMD-related analysis which has led to improved understandings of the illness’s medical manifestations, pure historical past, epidemiology, and genetics.
Ongoing work continues to give attention to FMD genetics and proteomics, physiological results of FMD on cardiovascular construction and perform, and novel imaging modalities and blood-based biomarkers that can be utilized to establish subclinical FMD. It can also be hoped that the subsequent decade will convey the event of multi-centred and probably worldwide medical trials to supply comparative effectiveness information to tell the optimum administration of sufferers with FMD. Notably, this research would possibly assist deal with preclinical vaccine assessments that at present make the most of primates as animal fashions, since autoimmune cross-reactions and the resultant hostile occasions can’t happen in absentia of shared sequences.
Immunohistochemistry and moleculargenetics within the differential diagnostics of mesenchymal lesions of gastrointestinal tract
Although, in routine observe, the differential diagnostics of mesenchymal tumors of the gastrointestinal tract continues to be targeted primarily on the proper analysis of gastrointestinal stromal tumor and its additional therapeutic administration based mostly on predictive diagnostics, latest progress within the improvement of endoscopic strategies has led to elevated detection of different mesenchymal lesions, which have been beforehand generally uncared for attributable to their small measurement or absence of signs requiring surgical exploration. Diagnosis of some of these lesions could also be reached based mostly on their histologic sample alone, whereas others could also be acknowledged with the use of tissue particular antibodies associated to the possible lineage of differentiation of the neoplastic cells.
Finally, a subset of tumors, generally with unsure lineage of differentiation, is outlined by pathognomonic genetic alterations of neoplastic cells. Recognition of such alterations, based mostly both on strategies of molecular genetics or immunohistochemical detection of an altered protein product, allows a exact analysis in a rising quantity of these instances. However, concerning the truth that most of these alterations will not be distinctive to a single tumor sort, however are sometimes shared by extra neoplastic entities, the analysis should nonetheless be based mostly on a posh diagnostic perspective, reflecting histological, immunohistochemical and molecular genetic options of the investigated tumor.
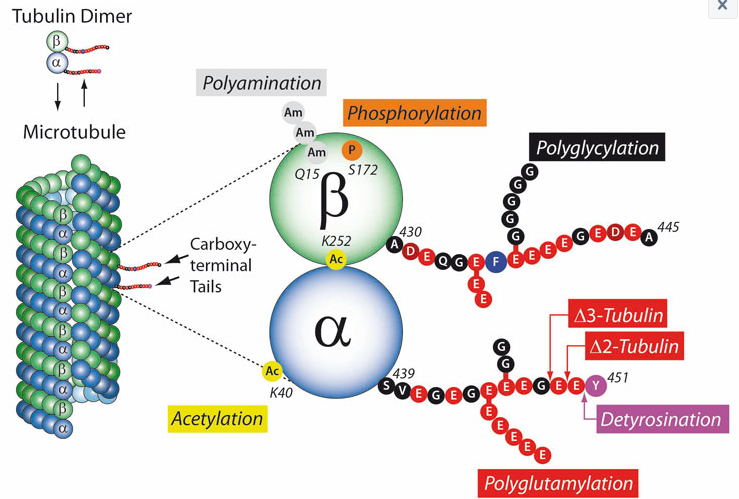
Post-translational modifications (PTMs) are highly dynamic and often reversible processes in which the functional properties of proteins are changed by adding chemical groups or other proteins to the amino acid residues. Tubulins and thus microtubules (MTs) are important target substrates for a large number of PTMs because they play a key role in cytoskeletal development and therefore play an important role in neuronal development, growth, cell motility and intracellular transport. The post-translational modifications include tyrosination or detyrosination, α2-tubulin formation, acetylation, phosphorylation, polyamination, ubiquitination, polyglutamylation and glycination (see figure). Most of these PTMs usually take place on tubulin subunits already built into microtubules.
The PTMs convey various properties:
Tubulin acetylation usually occurs with stable microtubules. Acetylation does not directly stabilize MTs but modifies the behavior of the proteins in the MT lumen.
Detyrosination of the C-terminal tyrosine of α-tubulin prevents the depolymerization of the microtubules and thereby increases their half-life.
Polyglutamylation, i.e. the formation of polyglutamate chains on the γ-carboxyl groups of glutamate residues is particularly pronounced during the differentiation of neuronal tissue. Polyglutamylation also regulates the stroke behavior of motile cilia by influencing the flagellar dynein motor. By activating microtubule-degrading enzymes such as spastine, polyglutamylation also stimulates MT turnover.
Tubulin polyglycination is the addition of glycine chains to the C-terminal domains of α- and β-tubulin. Polyglycination stabilizes the axonem – the central microtubule structure in cilia and flagella with the well-known 9×2 + 2 structure.
PTMs on microtubules generate a “tubulin code” that influences the biological functions of the MT cytoskeleton. The PTMs perform their function here by modulating higher MT structures and / or interactions with certain MT-associated proteins (MAPs, motor proteins, etc.). Microtubules are involved in various biological processes in practically every cell in the body. If this filigree regulated system is disturbed, this is an important factor in the development and clinical manifestation of Alzheimer’s, Parkinson’s and cancer.
Understanding cell death has a huge impact on the treatment of diseases. Apoptosis is a genetically determined suicide program that removes unnecessary or potentially harmful cells. Apoptosis is an important process in embryonic development, cell aging, the immune response and the response to poisoning. Deviations in this program can lead to neurodegenerative or autoimmune diseases. Blocking apoptosis is also one of the “hallmarks of cancer” postulated by Hanahan and Weinberg. 1
Apoptosis leads to a clear morphological change in the cell due to a non-inflammatory cascade of molecular events. These include the reduction of cell volume, the fragmentation of the cell nucleus, the condensation of chromatin, the degradation of DNA, the formation of bubbles on the cell membrane and the subsequent formation of apoptosis bodies. This sequence not only leads to the destruction of unwanted cells, but also prepares the cell debris for removal by phagocytes. In view of the variety of apoptotic stimuli and their effects on different signaling pathways, it is important to understand what exactly led to cell death in an experimental setup.
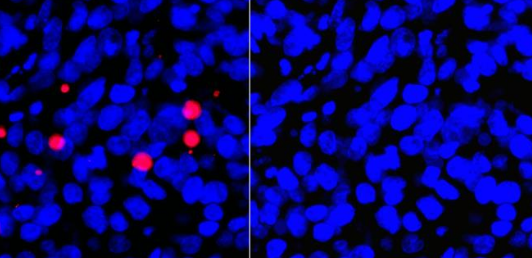
Mitochondrial membrane potential Mitochondria are involved in the apoptotic process in several places. During the life of a cell, mitochondria use oxidizable substrates to create a proton gradient along the inner mitochondrial membrane. During apoptosis, this membrane potential decreases in connection with the opening of the mitochondrial permeability pores and the release of apoptogenic factors such as cytochrome C. In some apoptotic models, the loss of membrane potential is considered an early event in the apoptotic process. Other researchers suspect that the loss of membrane potential is a result of the apoptotic signaling cascade. 2-4 An easy way to assess the membrane potential of mitochondria in a cell population is to use positively charged dyes such as JC-1 and TMRE, which are found in the electronegative Collect inside active mitochondria.
JC-1 / TMRE JC-1 changes from red fluorescence in healthy mitochondria to green fluorescence when, as in apoptosis, the membrane potential is lost. TMRE accumulates in polarized mitochondria and is particularly suitable for observing the membrane potential of living cells. Depolarized mitochondria with reduced membrane potential are unable to accumulate TMRE.
Caspase activation The key figures in the apoptotic signaling pathway are cysteine-dependent ASpartate-specific ProteASEN (caspases), the efficient activation of which determines the fate of the cell. Caspases are activated by several signal paths and form reinforcing loops. The external signaling pathway binds extracellular death ligands such as FasL or TNF-α to transmembrane death receptors, which in turn recruit initiator caspase-8. In the inner signal pathway, the cytochrome C released by mitochondria can trigger the formation of the caspase-activating complex (also called apoptosome). The latter in turn recruits and activates the initiator caspase-9. The initiator caspases then cleave and activate the effector caspases 3 and 7 and lead to the cleavage of specific substrates, which leads directly to the morphological changes that traditionally define cellular apoptosis.5 This is a measurement of the caspase activity important indicator of the ongoing apoptotic process.
Caspase 3/7 The protease activity of caspases 3 and 7 can be detected using the fluorogenic substrate N-Ac-DEVD-N’-MC-R110, which produces a fluorescent product on cleavage.
Core fragmentation, chromatin condensation and DNA degradation Caspases also break down internal cell structures and help with their efficient disposal. One of the most remarkable events in this process is the condensation of the cell nucleus and its breakdown into smaller fragments. The core fragmentation results from the dissolution of the nuclear lamina after proteolysis by caspases and the collapse of the nuclear envelope. Another characteristic of apoptosis is the condensation of chromatin, accompanied by hydrolysis of the core DNA. Hoechst 33342, a cell-permeable, fluorescent DNA dye, is often used to microscopically analyze chromatin condensation. The Golgi, the endoplasmic reticulum and the mitochondrial network also disintegrate during apoptosis. During the breakdown, the fragments are distributed in bubbles along the plasma membrane.
DNA dyes Hoechst dyes are cell-compatible and bind nucleic acids in living and fixed cells. Its blue, and therefore little overlapping, emission spectrum makes it recommended for researchers who plan to carry out several fluorescent stains on a sample. An alternative cell-compatible DNA dye is DRAQ5 ™, whose deep red excitation spectrum can be combined with fluorophores that emit blue and orange light. Non-membrane-compatible dyes such as propidium iodide, DAPI, DRAQ7 ™ or RedDot ™ 2 are ideal for the specific staining of the nuclei of dead cells where the integrity of the plasma membrane is impaired.
Blistering Caspases also cleave many major components of the cytoskeleton, thereby rounding and retracting the cells, which is typical of early stages of apoptosis. Another result of the weakening of the cytoskeleton is the formation of membrane vesicles. When the cytoplasm presses against weakened areas of the plasma membrane, these show themselves as bulges that can be visualized microscopically. These vesicles are believed to be a result of myosin-dependent contraction of cortical actin bundles that press the cytoplasm against the cell cortex. Blistering on the plasma membrane is an important step on the path of the apoptotic cell towards the smaller apoptotic bodies.
Elimination of apoptotic cells A critical component of the apoptotic process is the complete lack of an inflammatory response to dying cells. The disintegration of the apoptotic cells into apoptotic bodies prevents the release of the Damage-Associated Molecular Patterns (DAMPs) into the extracellular space and thereby facilitates the disposal by phagocytes. In early apoptosis, apoptotic cells secrete a “find me” signal and later a “eat me” signal to recruit phagocytes. This targeted interaction between apoptotic cells and phagocytes ensures that dying cells are eliminated by the non-inflammatory signaling pathway. “Find me” signals that recruit phagocytes include lysophosphatidylcholine, sphingosine-1-phosphate, fractal kinine and nucleotides such as ATP and UTP. These nucleotides are released through a caspase-mediated channel opening of the pannexin-1 (PANX1) channels.6 The caspase-dependent PANX1 channel opening also allows a small group of fluorescent monomeric cyanine dyes such as TO-PRO®-3 to flow in. In this way, the pannexin channels can be used to identify cells that send “find me” signals. 7
As soon as phagocytes approach an apoptotic cell, they recognize phosphatidylserine and phosphatidylethanolamine residues that come from inside the phospholipid double membrane and are now exposed on the cell surface. This rearrangement, which is typical of apoptotic cells, distinguishes it from its viable counterparts and provides the phagocytes with a “eat me” signal. The phospholipid-binding protein Annexin V adheres to phospholipid residues that are exposed during apoptosis. It is used to detect phosphatidylserine on the outer membrane of apoptotic cells and to determine the “eat me” phase of apoptosis.8,9
TO-PRO®-3 TO-PRO®-3 is a deep red fluorescent dye that enables the detection of an early event of apoptosis. The dye uses the caspase-dependent activated pannexin channels to penetrate the cells. This makes it possible to identify apoptotic cells that cannot be detected with the help of traditional markers such as Annexin V due to the process that has just started.
Annexin V Annexin V can be conjugated with various fluorochromes (e.g., FITC, PE, APC) to stain phosphatidylserine on the outer plasma membrane. The detection with Annexin V can be combined with other markers, such as those for membrane integrity (for example, RedDot ™ 2, propidium iodide, DAPI), in order to distinguish apoptotic from necrotic cells.
Multiplex is the key The above-mentioned morphological features of the apoptotic signaling pathway can help determine the type of cell death in a model system. However, a single measurement can lead to misinterpretations for certain experimental questions (e.g. the distinction between necrosis and apoptosis). In contrast to single measurements, multiplex assays provide a more complete picture of the apoptotic process. For example, the combined measurement with TO-PRO®-3, Annexin V, TMRE and DAPI allows a simultaneous statement on “Find me” or “Eat me” signals, the mitochondrial membrane potential and the core fragmentation. Multiplex assays thus provide a complex picture of the apoptotic processes and how they affect cells. In addition, markers for early apoptosis (eg TO-PRO®-3) can be used to identify cells that were considered viable due to the limitations of traditional apoptosis markers (eg Annexin V). Muliplex methods thus, by using different markers for the different phases of apoptosis, enable a quantitative classification of the entire cell population into the stages: viable, early apoptosis, late apoptosis, apoptosis bodies and non-cellular debris.
The selection of cell-based assays from Cayman Chemical provide you with flexible and efficient tools for determining the different stages of cell death in your model system. Biomol’s technical support and Cayman’s product developers will be happy to help you choose the cell-based assay that suits you, so that you are able to get the most accurate answers to your research questions.
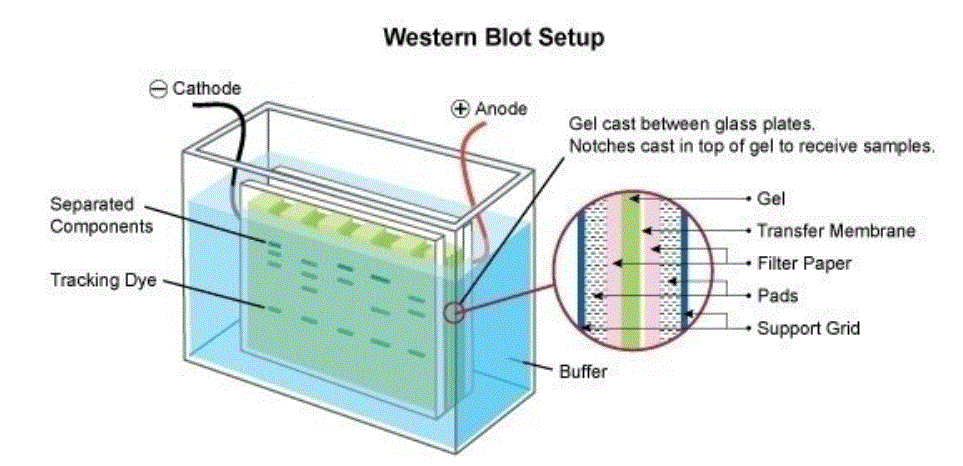
Western Blotting (also referred to as immunoblotting) is a technique used for analysis of individual proteins within a protein mix
(e.g. a cell lysate). In Western blotting (immunoblotting) the protein mix is applied to a gel electrophoresis at a carrier matrix (SDS-PAGE, native PAGE, isoelectric focusing, 2D gel electrophoresis, etc.) to sort the proteins by dimension , charge, or other differences in individual protein bands. The separated protein bands are then transferred to a carrier membrane (e.g.
nitrocellulose, nylon or PVDF). This process is known as blotting. As they’ve been separated due to interactions with fees, the proteins stick to the membrane in the exact same routine. The proteins within this immunoblot are then available for antibody binding for detection.
The antibodies are conjugated with fluorescent or radioactive tags or enzymes that give a subsequent response with an applied reagent, leading to a coloring or emission of light, enabling detection.
The term Western Blotting is based on a play of words. The southern blot, that can be a method to detect DNA sequences, is named after Ed Southern, who first described this process.
The western blot (immunoblot), in addition to the northern blot (for RNA detection), play the significance of this name.



![[Prospects for molecular-genetic support of research on proteolytics in the necrobiome composition]](https://osumolgen.org/wp-content/uploads/2021/03/IMG-20201217-WA0091-720x300.jpg "[Prospects for molecular-genetic support of research on proteolytics in the necrobiome composition]")
![[Prospects for molecular-genetic support of research on proteolytics in the necrobiome composition]](https://osumolgen.org/wp-content/uploads/2021/03/IMG-20201217-WA0091-300x142.jpg)
 Viruses in Sub-Saharan Africa from 2017 through 2019")